These are possible approaches to gaining a more systematic, enhanced understanding of the product and process under development.
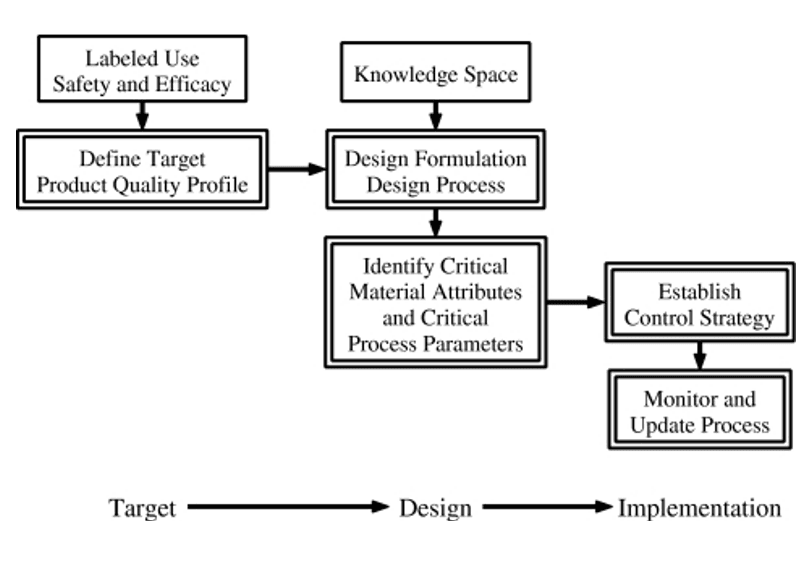
- Identify the critical process parameters and input (raw) material attributes that must be controlled to achieve these critical material attributes of the final product.
- Use risk assessment to prioritize process parameters and material attributes for experimental verification.
- Combine prior knowledge with experiments to establish a design space or other representation of process understanding.
- Establish a control strategy for the entire process that may include input material controls, process controls and monitors, design spaces around individual or multiple unit operations, and/or final product tests.
- The control strategy should encompass expected changes in scale and can be guided by a risk assessment.
- Continually monitor and update the process to assure consistent quality
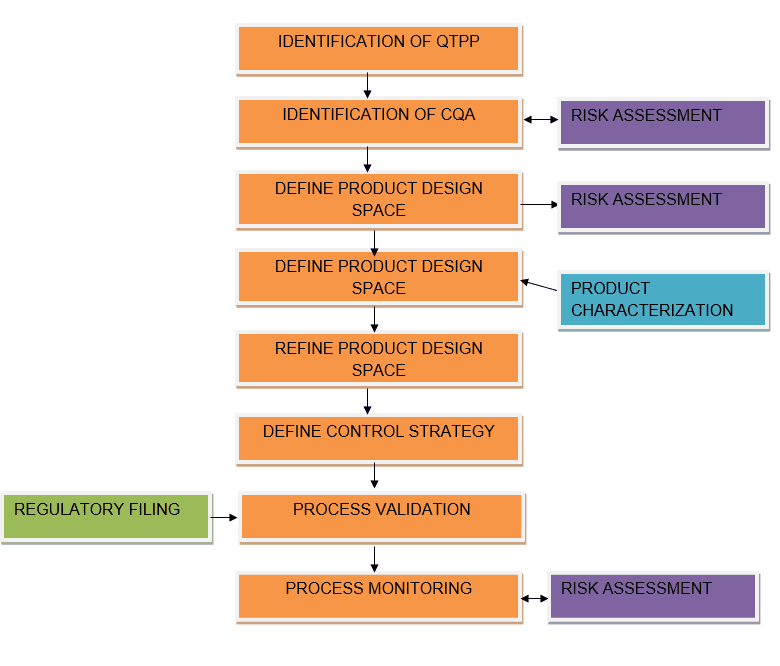
Quality Target Product Profile (QTPP):
A prospective summary of the quality characteristics of a drug product that ideally will be achieved to ensure the desired quality, taking into account safety and efficacy of the drug product.
The quality target product profile forms the basis of design for the development of the product. Considerations for the quality target product profile could include:
- Intended use in clinical setting, route of administration, dosage form, delivery Systems,
- Dosage strength,
- Container closure system,
- Therapeutic moiety release or delivery and attributes affecting pharmacokinetic characteristics (e.g., dissolution, aerodynamic performance) appropriate to the drug product dosage form being developed,
- Drug product quality criteria (e.g. purity, stability and drug release) appropriate for the intended marketed product.
- Provides an overall intent of the drug development program.
- QTPP is a patient and labeling centered concept, it can be thought of as the “user interface” of the drug product
- QTPP links drug development activities to specific concepts intended for inclusion in the drug labeling
Critical Quality Attributes:
A CQA is a physical, chemical, biological, or microbiological property or characteristic that should be within an appropriate limit, range, or distribution to ensure the desired product quality.
CQAs are generally associated with the drug substance, excipients, intermediates (in-process materials) and drug product. CQAs of solid oral dosage forms are typically those aspects affecting product purity, strength, drug release and stability. For drug substances, raw materials and intermediates, the CQAs can additionally include those properties (e.g., particle size distribution, bulk density) that affect drug product CQAs. Potential drug product CQAs derived from the quality target product profile and/or prior knowledge is used to guide the product and process development. The list of potential CQAs can be modified when the formulation and manufacturing process are selected and as product knowledge and process understanding increase. Quality risk management can be used to prioritize the list of potential CQAs for subsequent evaluation. Relevant CQAs can be identified by an iterative process of quality risk management and experimentation that assesses the extent to which their variation can have an impact on the quality of the drug product.
Critical Material Attributes (CMA – API & Excipients):
A physical, chemical, biological or microbiological property or characteristic of a material that should be within an appropriate limit, range, or distribution to ensure the desired product quality
The manufacturing process development program should identify which material attributes (e.g., of raw materials, starting materials, reagents, solvents, process aids, intermediates) and process parameters should be controlled. Risk assessment can help identify the material attributes and process parameters with the potential for having an effect on drug substance CQAs. Those material attributes and process parameters that are found to be important to drug substance quality should be addressed by the control strategy.
The risk assessment used to help define the elements of the control strategy that pertain to materials upstream from the drug substance can include an assessment of manufacturing process capability, attribute detectability, and severity of impact as they relate to drug substance quality. For example, when assessing the link between an impurity in a raw material or intermediate and drug substance CQAs, the ability of the drug substance manufacturing process to remove that impurity or its derivatives should be considered in the assessment. The risk related to impurities can usually be controlled by specifications for raw material/intermediates and/or robust purification capability in downstream steps. The risk assessment can also identify CQAs for which there are inherent limitations in detectability in the drug substance (e.g., viral safety). In these cases, such CQAs should be controlled at an appropriate point upstream in the process.
For chemical entity development, a major focus is knowledge and control of impurities. It is important to understand the formation, fate (whether the impurity reacts and changes its chemical structure), and purge (whether the impurity is removed by, for example, crystallization, extraction), as well as their relationship to the resulting impurities that end up in the drug substance as CQAs. The process should be evaluated to establish appropriate controls for impurities as they progress through multiple process operations.
Using a traditional approach, material specifications and process parameter ranges can be based primarily on batch process history and univariate experiments. An enhanced approach can lead to a more thorough understanding of the relationship of material attributes and process parameters to CQAs and the effect of interactions. Example 1 illustrates the development of process parameters using prior knowledge and chemistry-first principles.
Risk assessment can be used during development to identify those parts of the manufacturing process likely to have an impact on potential CQAs. Further risk assessments can be used to focus development work on areas for which better understanding of the link between process and quality is needed. Using an enhanced approach, the determination of appropriate material specifications and process parameter ranges could follow a sequence such as the one shown below: Identify potential sources of process variability.
- Identify the material attributes and process parameters likely to have the greatest impact on drug substance quality. This can be based on prior knowledge and risk assessment tools.
- Design and conduct studies (e.g., mechanistic and/or kinetic evaluations, multivariate design of experiments, simulations, modelling) to identify and confirm the links and relationships of material attributes and process parameters to drug substance CQAs.
- Analyze and assess the data to establish appropriate ranges, including establishment of a design space if desired.
- Small-scale models can be developed and used to support process development studies. The development of a model should account for scale effects and be representative of the proposed commercial process. A scientifically justified model can enable a prediction of quality, and can be used to support the extrapolation of operating conditions across multiple scales and equipment.
Risk Assessment: Linking Material Attributes and Process Parameters to Drug Product CQAs:
Risk assessment is a valuable science-based process used in quality risk management that can aid in identifying which material attributes and process parameters potentially have an effect on product CQAs. Risk assessment is typically performed early in the pharmaceutical development process and is repeated as more information becomes available and greater knowledge is obtained.
Risk assessment tools can be used to identify and rank parameters (e.g., process, equipment, input materials) with potential to have an impact on product quality, based on prior knowledge and initial experimental data. The initial list of potential parameters can be quite extensive, but can be modified and prioritized by further studies (e.g., through a combination of design of experiments, mechanistic models). The list can be refined further through experimentation to determine the significance of individual variables and potential interactions. Once the significant parameters are identified, they can be further studied (e.g., through a combination of design of experiments, mathematical models, or studies that lead to mechanistic understanding) to achieve a higher level of process understanding.
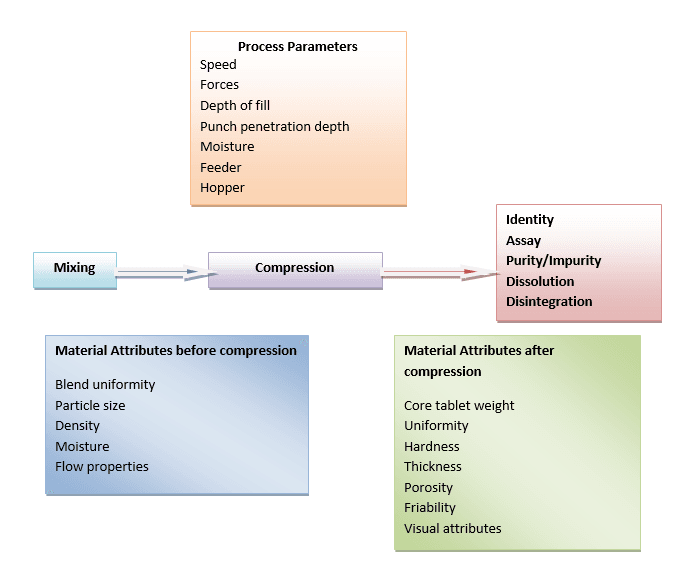
Critical Process Parameters (CPP):
- Critical process parameter (CPPs): the input operating parameters (mixing speed, flow rate) and process state variables (temperature, pressure) of a process or unit operation
- For a given unit operation, there are four categories of parameters and attributes
- input material attributes
- output material attributes
- input operating parameters
- output process state conditions
- the state of a process depends on its CPPs and the CMAs of the input materials
- monitoring and controlling output material attributes can be a better control strategy than monitoring operating parameters especially for scale up
- a material attribute, such as moisture content, should have the same target value in the pilot and commercial processes.
- an operating parameter, such as air flow rate, would be expected to change as the process scale changes.
- Different sets of CPP can have several origins.
- definition of operating parameters depends on the engineering systems installed on a process equipment
- e.g., one fluid bed dryer may define the product temperature as an operating parameter (a thermostat maintaining that temp.) while another fluid bed dryer may have inlet air flow rate & inlet air temperature indicated as operating parameters
- batch record for the first unit might indicate a fixed temperature, while the second unit would have a design space that indicated the combination of inlet air flow rate and inlet air temperature that would insure the appropriate product temperature
- differences in the set of CPP comes from the balance between control of operating parameters and material attributes
- in fact, a set of CPP and CMA (which he refers to as process critical control points (PCCP)) can affect the scale up process
- definition of operating parameters depends on the engineering systems installed on a process equipment
Control Strategy:
A control strategy is designed to ensure that a product of required quality will be produced consistently. The elements of the control strategy should be described and justified how in-process controls and the controls of input materials (drug substance and excipients), intermediates (in-process materials), container closure system, and drug products contribute to the final product quality.
These controls should be based on product, formulation and process understanding and should include, at a minimum, control of the critical process parametersand material attributes.
A comprehensive pharmaceutical development approach will generate process and product understanding and identify sources of variability. Sources of variability that can impact product quality should be identified, appropriately understood, and subsequently controlled. Understanding sources of variability and their impact on downstream processes or processing, in-process materials, and drug product quality can provide an opportunity to shift controls upstream and minimise the need for end product testing. Product and process understanding, in combination with quality risk management will support the control of the process such that the variability (e.g., of raw materials) can be compensated for in an adaptable manner to deliver consistent product quality.
This process understanding can enable an alternative manufacturing standard where the variability of input materials could be less tightly constrained. Instead it can be possible to design an adaptive process step (a step that is responsive to the input materials) with appropriate process control to ensure consistent product quality. Enhanced understanding of product performance can justify the use of alternative approaches to determine that the material is meeting its quality attributes. The use of such alternatives could support real time release testing. For example, disintegration could serve as a surrogate for dissolution for fast-disintegrating solid forms with highly soluble drug substances. Unit dose uniformity performed in-process (e.g., usingweight variation coupled with near infrared (NIR) assay) can enable real time release testing and provide an increased level of quality assurance compared to the traditional end-product testing using compendial content uniformity standards. Real time release testing can replace end product testing, but does not replace the review and quality control steps called for under GMP to release the batch.
A control strategy can include, but is not limited to, the following:
- Control of input material attributes (e.g., drug substance, excipients, primary packaging materials) based on an understanding of their impact on processability or product quality;
- Product specification(s);
- Controls for unit operations that have an impact on downstream processing or product quality (e.g., the impact of drying on degradation, particle size distribution of the granulate on dissolution);
- In-process or real-time release testing in lieu of end-product testing (e.g. measurement and control of CQAs during processing);
- A monitoring program (e.g., full product testing at regular intervals) for verifying multivariate prediction models.
A control strategy can include different elements. For example, one element of the control strategy could rely on end-product testing, whereas another could depend on real-time release testing. The rationale for using these alternative approaches should be described in the submission.
Adoption of the principles in this guideline can support the justification of alternative approaches to the setting of specification attributes and acceptance criteria as described in Q6A and Q6B.
The classification of process parameters as critical or non-critical is essential to evolve the control strategy toward the QbD based goal. Full classification of all parameters as either non-critical or critical can lead to reduced end-product testing. It is the uncertainty about the UPP that leads to extensive testing.
Without development studies, UPP may need to be constrained at fixed values or narrow ranges (used to produce acceptable exhibit batches) because they might be critical. The presence of UPP also leads to inclusion of extensive release and in-process tests into the control strategies. The goal of development studies is to move parameter from unclassified (criticality unknown) to either non-critical or critical. This classification is an important step toward a flexible manufacturing process because unclassified parameters classified as non-critical may be monitored and controlled via monovarient ranges or as part of a sponsor’s quality system. For non-critical parameters it may be possible to designate a normal operation range (NOR) up to (or beyond) the proven acceptable range (PAR) depending on trends and prior knowledge. The superposition of NOR for non-critical parameters would be considered as part of the design space.
The ranges of critical parameters must be constrained to a multidimensional design space or fixed at values of all parameters known to be acceptable. Univariate PAR can be used for critical parameters only when there is evidence that there are no significant interactions between the CPP. However, the establishment of this knowledge about CPPs may render them lower risk than UPP. A control strategy appropriate to the known CPP may also have less need for release testing than one for a process with many UPPs.