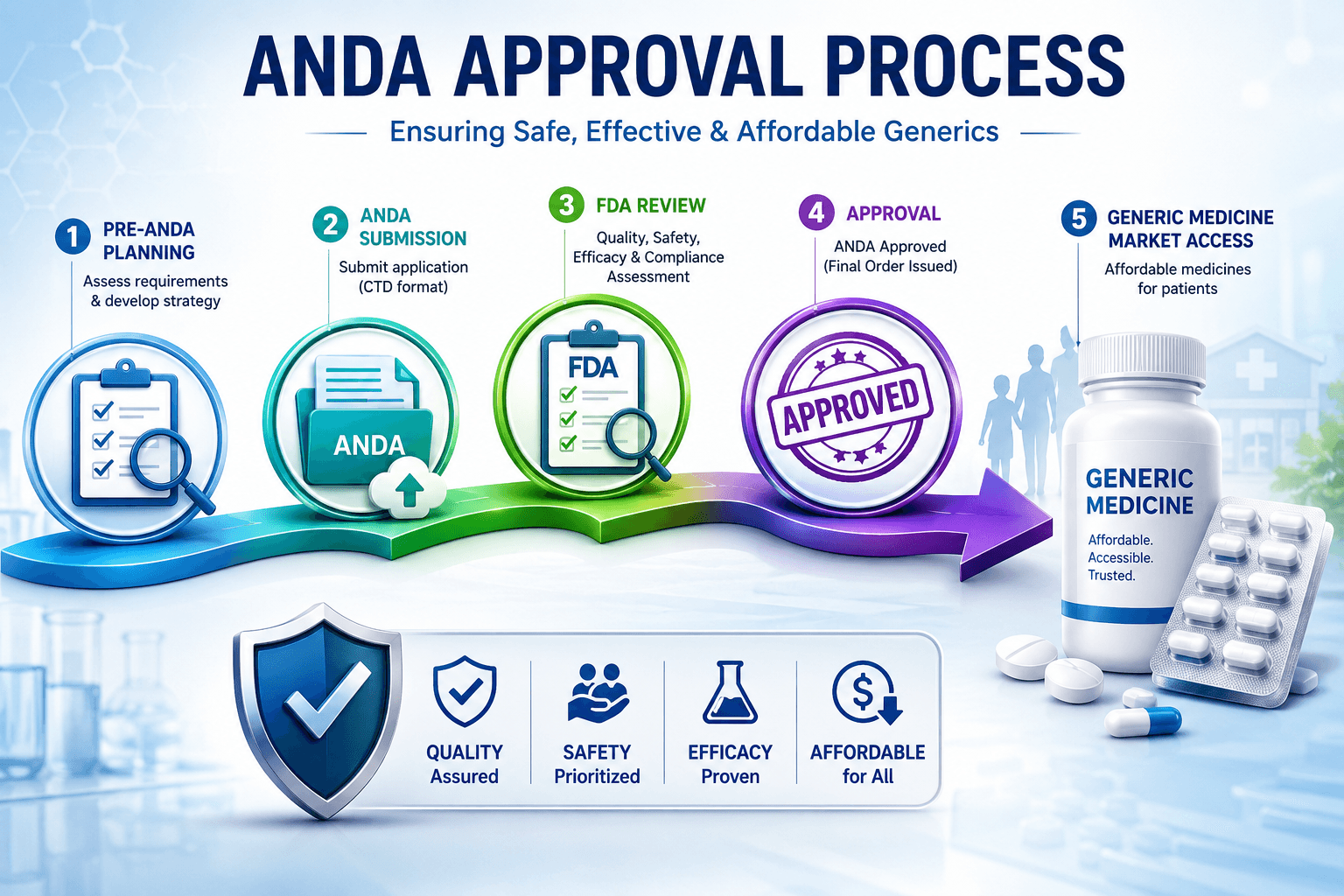
ANDA Approval Process in USFDA: Complete Guide for Generic Drug Approval 2026
Want to launch a generic drug in USA? You need ANDA – Abbreviated New Drug Application approval from USFDA. Unlike NDA for innovators, ANDA proves your generic is same as Reference Listed Drug RLD in safety + efficacy.
This guide covers ANDA vs NDA, step-by-step ANDA submission process, DMF linkage, bioequivalence BE studies, GDUFA fees, USFDA review timeline, and top reasons ANDAs get Complete Response Letter CRL in 2026.
1. What is ANDA – Abbreviated New Drug Application
Definition per 21 CFR 314.94: ANDA is application for USFDA approval to market a generic drug product. “Abbreviated” because you don’t repeat costly safety/efficacy clinical trials done by innovator.
Key Concept: You prove “sameness” to RLD = Reference Listed Drug. Same active ingredient, dosage form, route, strength, and bioequivalence.
ANDA vs NDA vs 505(b)(2):
| Application | For | Clinical Data Needed | Review Time Target |
|---|---|---|---|
| NDA 505(b)(1) | New innovator drug | Full safety + efficacy trials | 10 months standard |
| ANDA 505(j) | Generic of approved drug | Only Bioequivalence BE data | 10 months GDUFA goal |
| 505(b)(2) | Modified innovator | Partial clinical data | 10 months standard |
2. Prerequisites Before ANDA Submission
- Identify RLD: Check USFDA Orange Book for Reference Listed Drug + therapeutic equivalence code
- Patent & Exclusivity Check: File Paragraph I-IV certification. Para IV = challenge patent = chance for 180-day exclusivity
- DMF Availability: API supplier must have USFDA accepted DMF Type II. You’ll reference it in ANDA
- GMP Compliance: Manufacturing site must pass USFDA pre-approval inspection PAI. No 483s pending
- BE Study: Conduct bioequivalence study vs RLD in USFDA approved CRO. Fasting + fed study usually required
3. ANDA Submission Steps – 8 Stages
Step 1: Development & Formulation
Develop generic formulation matching RLD. Use QbD approach per ICH Q8. Characterize CQA, CPP. Do forced degradation, compatibility studies.
Step 2: DMF Linkage
API vendor files DMF Type II with USFDA. You get “Letter of Authorization LOA” to reference their DMF. Without DMF, ANDA will be refused.
Step 3: Bioequivalence BE Studies
Conduct single-dose, crossover BE study vs RLD. 90% CI for Cmax, AUC must be 80-125%. Study must follow USFDA product-specific guidance PSG.
Step 4: ANDA Dossier Preparation
Submit in eCTD format via ESG gateway. Module 1-5 structure same as NDA but Module 4 clinical section is replaced by BE data.
Module 3 – CMC: Drug substance, drug product, manufacturing process, process validation, stability data – 3 months accelerated + 6 months long term min at submission
Step 5: GDUFA Fee Payment
Pay USFDA GDUFA fee before submission. FY2026 fee ~ $813,936 for ANDA. Small business gets waiver. No fee = RTR – Refuse to Receive.
Step 6: ANDA Submission & RTR Check
Submit via ESG. USFDA does 60-day RTR check. If dossier incomplete, miss GDUFA fee, or wrong eCTD format = RTR. Fix and resubmit.
Step 7: USFDA Review + PAI
GDUFA III Goal: 10 months for standard review, 8 months for priority. During review:
- USFDA asks Information Request IR – 30 days to reply
- Discipline Review Letter DRL issued if major issues
- Pre-Approval Inspection PAI of API + FDF site by USFDA investigator
Step 8: Approval or Complete Response Letter CRL
If everything compliant = ANDA Approval. If issues = CRL listing deficiencies. You get 1 year to respond. After approval, you can launch post patent/exclusivity expiry.
4. USFDA Review Timeline – GDUFA III Commitments
| Milestone | Timeline | Notes |
|---|---|---|
| RTR Check | 60 days | Complete dossier check |
| Standard Review | 10 months | From submission date |
| Priority Review | 8 months | For drug shortage/competitive generic |
| CRL Response | 10 months | Clock restarts after your reply |
Actual time often 14-24 months due to IR, DRL, and PAI scheduling delays.
5. Top 7 Reasons ANDAs Get CRL in 2026
- Bioequivalence issues: 90% CI outside 80-125%, wrong PSG followed, CRO not USFDA inspected
- CMC deficiencies: Impurity profile not matching RLD, stability data gap, container closure issue
- Manufacturing facility 483: PAI finds data integrity, sterility, or process control issues
- DMF not adequate: API DMF has deficiency, USFDA puts it on “Hold”
- Labeling mismatch: Patient labeling not same as RLD per 21 CFR 314.94(a)(8)
- Patent certification error: Wrong Para IV notice sent to innovator
- Facility not ready: Site added in ANDA but not inspected/approved yet
6. Post-Approval: What After ANDA Approval
- Launch: Can market only after patent + exclusivity expiry. Para IV filer gets 180-day exclusivity
- Post-approval changes: CBE-30, CBE-0, PAS supplements per 21 CFR 314.70
- Annual Report: Submit yearly update on manufacturing changes
- GDUFA facility fees: Pay every year for FDF + API site to stay in USFDA list
Key Takeaway for Regulatory Affairs
ANDA approval is 70% science + 30% regulatory strategy. 3 golden rules:
- BE study is king: One failed BE = 6 months + $1M lost. Use USFDA approved CRO only
- DMF + Site readiness: Don’t file ANDA if DMF or PAI is pending
- QbD dossier: USFDA gives fewer IR if you show Design Space + Control Strategy per ICH Q8/Q11
First ANDA approval is hardest. Once USFDA trusts your site + dossier quality, subsequent ANDAs move faster.
Regulatory Disclaimer
This article is for educational purposes for pharma regulatory affairs and QA professionals. ANDA submission must follow 21 CFR 314, USFDA GDUFA III guidance, and current Orange Book. Patent strategy requires legal counsel. USFDA review timelines are goals, not guarantees. This is not legal or regulatory consultancy.

About the Author
Mahummed Asif is a pharmaceutical QA professional with 16 years experience in GMP, Process Validation, QMS, and regulatory audits. Pharmashare.in content references 21 CFR 314, ICH Q8/Q11, and USFDA GDUFA guidance as of April 2026.
Need ANDA Submission Checklist Excel? Contact Pharmashare.in for free template.
Excellent information asif sir!
We need pdf to easy download sir
Share your e-mail address, will share you
Very nice and very informative contents.
Very nice and very informative contents…
Thank you, Mahummed Asif, for the valuable information
Could you please share the PDF link with me at this email address: halhewsheh@hikma.com.