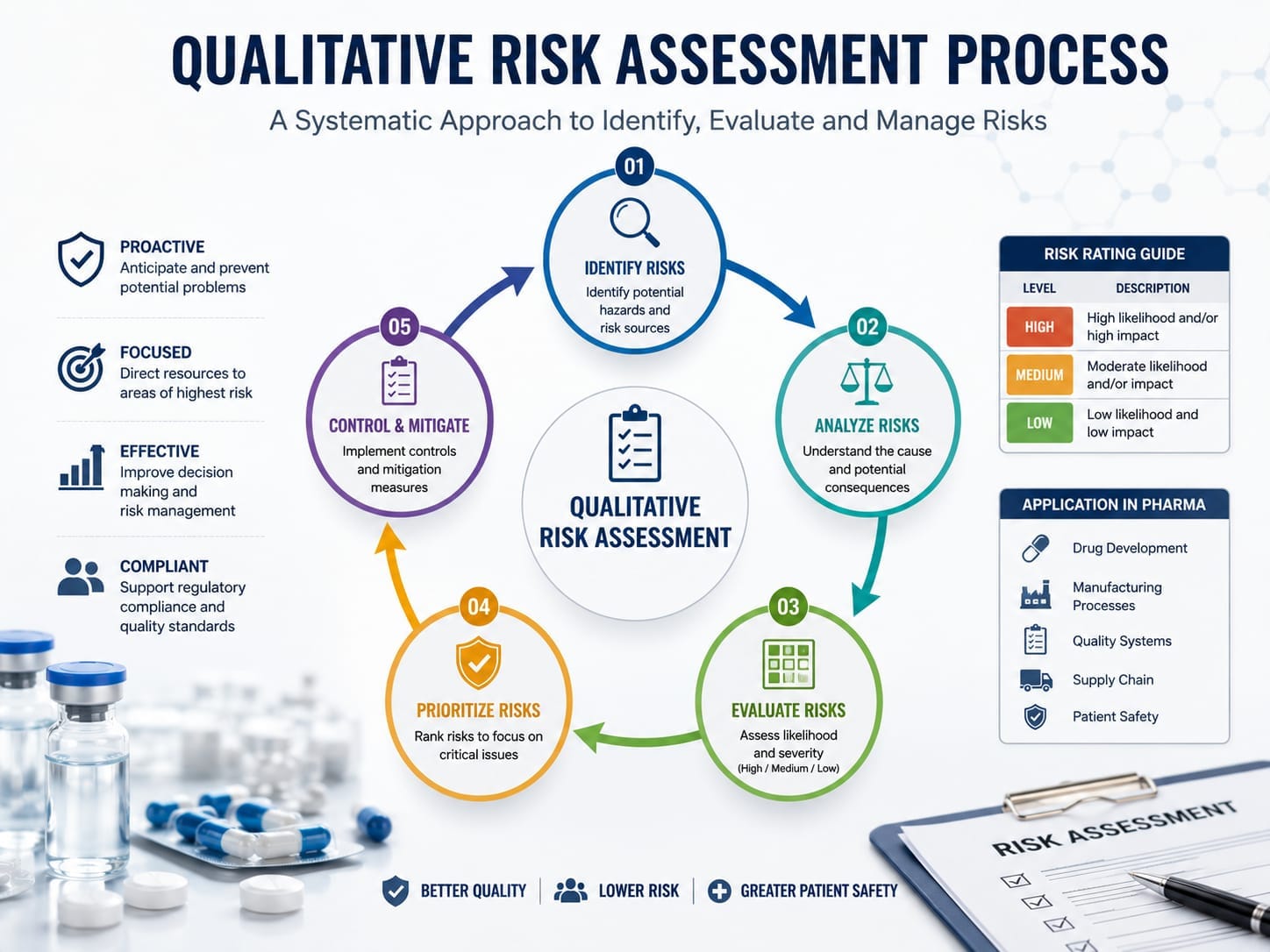
Qualitative Risk Assessment Process
A qualitative risk assessment should follow a structured and documented process to ensure consistency, transparency, and regulatory compliance. Although organizations may customize their approach, the methodology generally aligns with the Quality Risk Management (QRM) framework described in ICH Q9(R1).
Step 1 – Define the Scope
Clearly define the objective of the assessment before identifying risks. The scope should specify:
- Product or process being evaluated
- Purpose of the assessment
- Applicable regulatory requirements
- Departments involved
- Assessment boundaries
Example:
“Evaluate the impact of replacing the tablet compression machine on product quality, validation status, and regulatory compliance.”
Step 2 – Identify Hazards
The cross-functional team identifies every possible hazard that could affect product quality, patient safety, or regulatory compliance.
Typical hazards include:
- Equipment malfunction
- Incorrect dispensing
- Operator error
- Cross contamination
- Microbial contamination
- Incorrect documentation
- Label mix-up
- Power failure
- Incorrect cleaning
- Data integrity issues
- Supplier quality failures
- Environmental excursions
Step 3 – Analyze the Risk
The team evaluates each hazard based on available scientific knowledge, historical trends, investigations, validation data, audit observations, and process understanding.
Unlike quantitative methods, qualitative assessment does not require mathematical calculations. Instead, descriptive ratings are assigned using predefined criteria.
| Risk Factor | Typical Rating |
|---|---|
| Likelihood | Low / Medium / High |
| Severity | Minor / Moderate / Major / Critical |
| Detectability (Optional) | Easy / Moderate / Difficult |
Step 4 – Evaluate the Risk
After identifying and analyzing hazards, each risk is categorized according to predefined acceptance criteria.
| Risk Level | Recommended Action |
|---|---|
| High | Immediate mitigation required |
| Medium | Risk reduction recommended |
| Low | Accept with periodic review |
Step 5 – Implement Risk Controls
Appropriate controls should reduce either the likelihood of occurrence, the severity of impact, or improve detectability.
Examples include:
- Additional operator training
- Equipment qualification
- Process validation
- Preventive maintenance
- Additional in-process controls
- Automated alarms
- Electronic batch records
- Improved SOPs
- Independent verification
- Supplier qualification improvements
Step 6 – Review Risk
Risk assessments should never remain static. They should be periodically reviewed whenever significant changes occur, including:
- Process changes
- Regulatory updates
- Customer complaints
- Audit observations
- Trend analysis
- Deviation recurrence
- Annual Product Quality Review (APQR)
Qualitative Risk Matrix
The risk matrix is one of the simplest and most effective qualitative tools. It combines the probability of occurrence with the severity of impact to determine the overall level of risk.
| Severity ↓ / Likelihood → | Low | Medium | High |
|---|---|---|---|
| Critical | Medium | High | High |
| Major | Low | Medium | High |
| Moderate | Low | Medium | Medium |
| Minor | Low | Low | Medium |
Organizations may customize this matrix according to their Pharmaceutical Quality System, but the rationale should remain consistent across assessments.
Practical Pharmaceutical Examples
Example 1 – Change Control
| Assessment Item | Evaluation |
|---|---|
| Change | New tablet press installation |
| Potential Risk | Compression force variation |
| Likelihood | Medium |
| Severity | High |
| Overall Risk | High |
| Mitigation | Qualification, validation, operator training |
Example 2 – Cleaning Validation
| Assessment Item | Evaluation |
|---|---|
| Hazard | Residue carryover |
| Likelihood | Low |
| Severity | Critical |
| Overall Risk | Medium |
| Mitigation | Validated cleaning procedure and periodic monitoring |
Example 3 – Supplier Qualification
| Assessment Item | Evaluation |
|---|---|
| Risk | Raw material variability |
| Likelihood | Medium |
| Severity | Major |
| Overall Risk | Medium |
| Mitigation | Supplier audit, incoming testing, quality agreement |
Qualitative vs Quantitative Risk Assessment
| Parameter | Qualitative | Quantitative |
|---|---|---|
| Data Required | Limited | Extensive |
| Uses Numerical Scores | No | Yes |
| Expert Judgment | High | Moderate |
| Speed | Fast | Time-consuming |
| Best For | Routine GMP decisions | Complex engineering analysis |
| Ease of Implementation | Very Easy | Moderate to Difficult |
Advantages of Qualitative Risk Assessment
- Simple to understand and implement
- Requires minimal statistical knowledge
- Supports rapid decision-making
- Ideal for cross-functional discussions
- Promotes proactive quality culture
- Widely accepted by regulatory agencies
- Suitable for small and large pharmaceutical organizations
- Cost-effective compared to complex quantitative models
Limitations
- Depends heavily on expert judgment
- May introduce subjective bias
- Different teams may assign different ratings
- Less precise than quantitative methods
- Not suitable for highly complex engineering risk calculations
Coming in Part 3
- Best practices for qualitative risk assessments
- Common mistakes observed during FDA and EU inspections
- Documentation requirements
- Risk assessment template
- Frequently Asked Questions (FAQs)
- Conclusion
- Author bio
- Regulatory disclaimer
Author
Mahummed Asif
Mahummed Asif is a pharmaceutical quality professional and the founder of PharmaShare. He specializes in Pharmaceutical Quality Assurance, Regulatory Affairs, Validation, GMP compliance, and global regulatory guidelines. Through PharmaShare, he publishes practical resources, regulatory updates, and technical articles to help pharmaceutical professionals strengthen compliance and enhance their industry knowledge.
Regulatory Disclaimer
This article is intended solely for educational and informational purposes. While every effort has been made to ensure technical accuracy, readers should always refer to the latest versions of applicable regulations and official guidance documents, including ICH Q9(R1), US FDA guidance, EU GMP, WHO Technical Reports, and PIC/S publications before making regulatory or quality decisions. Organizational procedures and local regulatory requirements may differ.