USFDA Inspection Systems in Pharma: 6 Systems FDA Checks First + 2026 Survival Guide
Updated: April 19, 2026 | Category: Regulatory Compliance | Reading Time: 10 min
USFDA inspection is not an audit. Audit = you control timing. Inspection = FDA controls everything.
From FY 2023-2025 data, 68% of USFDA 483s trace back to just 2 systems: Quality System + Laboratory Controls. If you fix these, you fix 70% of inspection risk.
This guide breaks down all 6 GMP systems FDA inspectors evaluate, what they ask in first 4 hours, and how to stay 483-free in 2026.
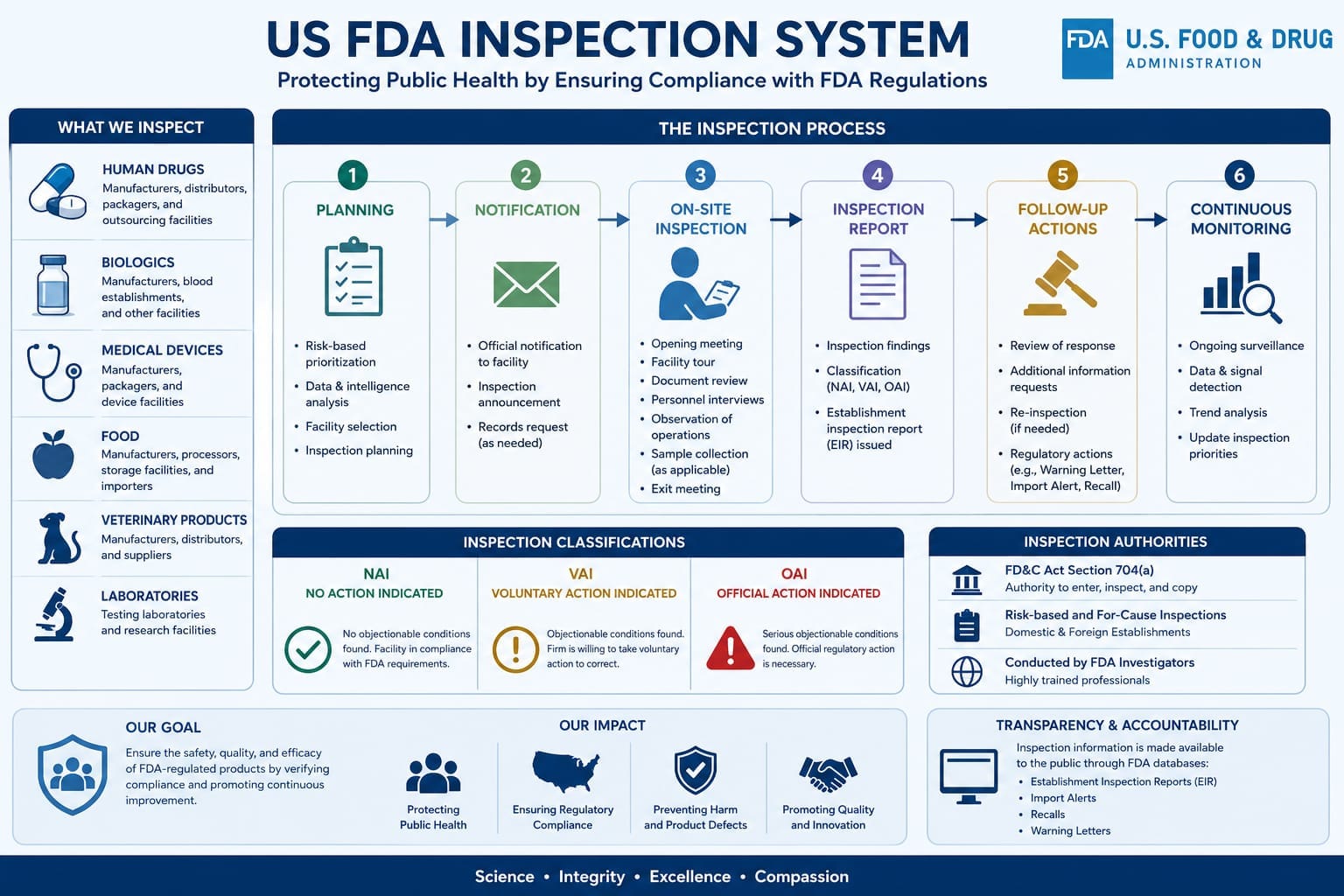
1. What Are USFDA “Systems” and Why They Matter
FDA doesn’t inspect “departments”. They inspect “systems” per Compliance Program 7356.002. Each system = people + procedures + data + equipment.
FDA Logic: “A weakness in Quality System usually means weakness in other 5 systems too.” – FDA Investigator Manual
2. The 6 GMP Systems FDA Evaluates
| System | What FDA Checks First | Top 483 Area 2024-25 |
|---|---|---|
| 1. Quality System | Management review, CAPA effectiveness, deviation trends, annual product review | Inadequate CAPA + No trending |
| 2. Production System | Line clearance, batch record review, equipment status, in-process testing | Batch record discrepancies |
| 3. Laboratory Controls | Data integrity, OOS/OOT investigations, method validation, audit trail review | Data integrity + retesting |
| 4. Materials System | Supplier approval, incoming sampling, quarantine/release, water system | Unapproved supplier use |
| 5. Facilities & Equipment | HVAC qualification, equipment qualification, maintenance, cleaning validation | No cleaning validation |
| 6. Packaging & Labeling | Line clearance, label control, reconciliation, tamper-evident features | Label mix-up risk |
FDA’s entry strategy 2026: Day 1 = Quality System + Lab Controls. If these 2 are clean, inspection depth reduces. If messy, they drill into Production + Materials for 10 days.
3. 6 Types of USFDA Inspections You Must Know
- Pre-Approval Inspection PAI: For ANDA/NDA. Focus = data integrity + process validation. 5-7 days.
- Surveillance/Biennial: Routine GMP. Every 2 years for US sites, risk-based for foreign. Checks all 6 systems.
- For-Cause Inspection: Triggered by complaint, recall, data fraud. Very deep, very fast.
- Compliance Follow-up: After Warning Letter. Only checks if WL commitments are fixed.
- CGMP + Pre-license Combo: For new sterile facilities. Longest – 12-14 days.
- Remote Records Review: Post-COVID trend. FDA asks eBPR, audit trails via secure portal before physical visit.
4. First 4 Hours: What FDA Asks Immediately
First impression = 50% of inspection tone. Prepare this “Day 1 Kit”:
- Org chart + site master file: Who’s responsible for each system
- Last 2 years 483 + response: Show CAPA closure evidence
- Quality metrics dashboard: Deviation rate, OOS rate, CAPA overdue. FDA loves metrics.
- Data integrity policy + audit trail SOP: 2026 hot topic due to vendor CSV issues
- List of critical equipment + qualification status: Sterilizers, HPLC, compression machines
Pro tip: Assign 1 “system owner” per 6 systems. FDA hates “Sir is on leave” answers.
5. 7 Walk-Through Mistakes = Instant 483
From recent India-site WLs, avoid these:
- “We’ll show you later”: If FDA asks for SOP, show within 10 min. Delay = suspicion.
- Escorting >3 people: 1 SME + 1 QA escort max. Crowd = nervousness.
- Correcting on the spot: Don’t back-date logbooks during walk-through. FDA watches hands.
- “I don’t know” from SME: If operator doesn’t know SOP step, FDA writes “inadequate training”.
- Open laptops during inspection: FDA will ask to see screen. Close non-GMP work.
- No water/area access: Blocking FDA from water room = automatic deep dive.
- Discussing other clients: “Our US client does this…” = breach of confidentiality + 483.
