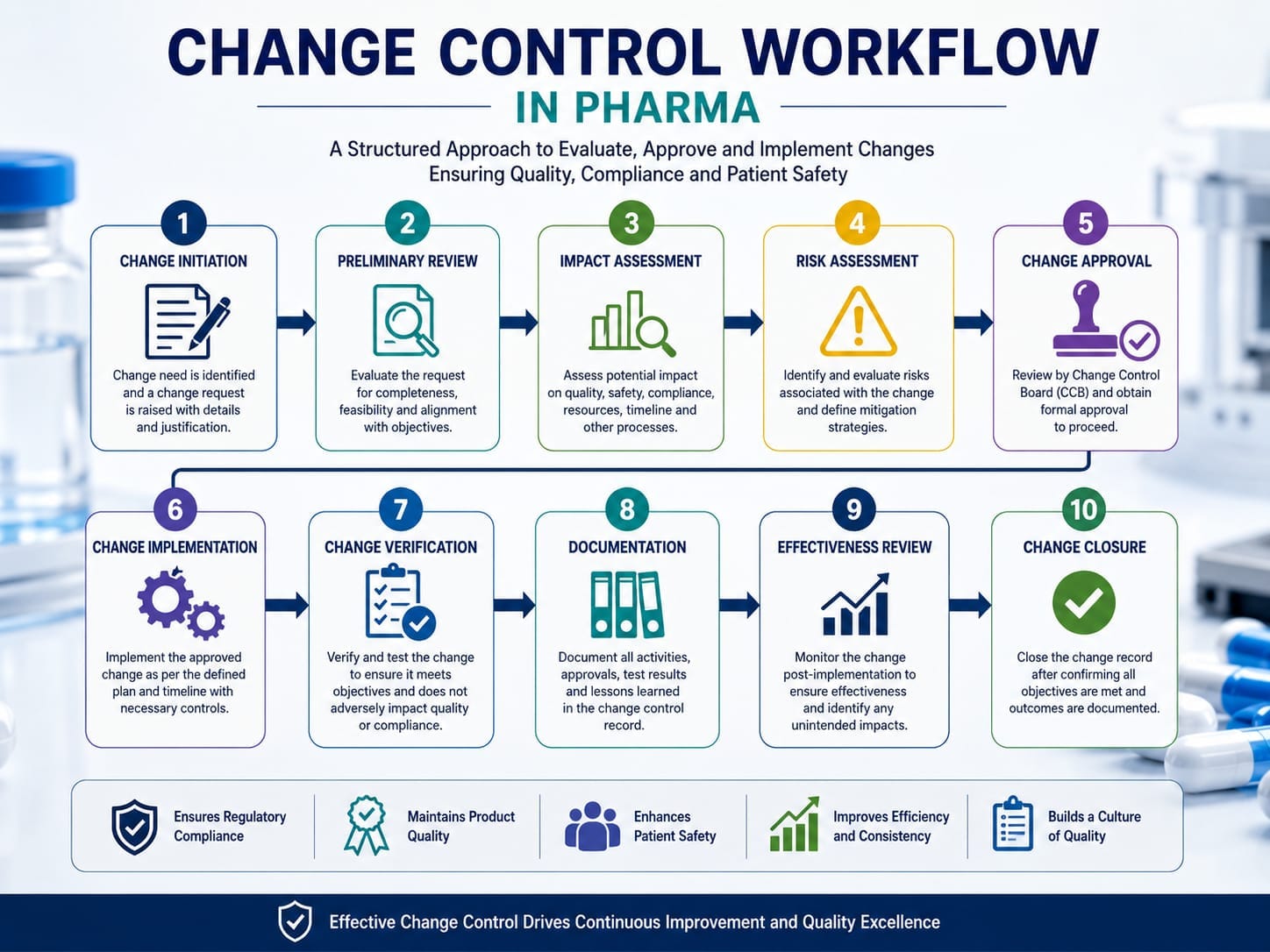
Complete Change Control Workflow in Pharmaceutical Industry
An effective Change Management System follows a structured workflow that ensures every proposed modification is scientifically evaluated before implementation. Although individual pharmaceutical companies may customize their procedures, the fundamental workflow remains largely consistent across the industry.
Step 1: Change Request Initiation
The process begins when a need for change is identified. The initiator documents the proposed change using the organization’s Change Control Form or Electronic Quality Management System (eQMS).
The request should include:
- Title of the proposed change
- Unique Change Control Number
- Reason for change
- Current process description
- Proposed modification
- Business justification
- Departments affected
- Expected implementation timeline
- Supporting documents
Step 2: Preliminary Review
The Quality Assurance department performs an initial review to determine whether:
- The change request is complete.
- The proposed change is clearly defined.
- Sufficient supporting information is available.
- The request falls within the scope of Change Control.
- Additional technical information is required.
If the proposal is incomplete, QA returns it to the initiator for clarification before proceeding.
Step 3: Quality Risk Assessment
Risk assessment is one of the most important stages of Change Management. The objective is to identify and evaluate potential risks associated with implementing the proposed change.
Typical questions include:
- Will product quality be affected?
- Could patient safety be compromised?
- Will the validated state be impacted?
- Does the change introduce contamination risks?
- Will regulatory filings require updates?
- Does the change affect data integrity?
- Could the change increase deviation rates?
Common Risk Assessment Tools
| Risk Tool | Typical Use |
|---|---|
| FMEA (Failure Mode and Effects Analysis) | Equipment, manufacturing and process changes |
| Risk Ranking | General quality risk evaluation |
| HACCP | Contamination control |
| Fishbone Diagram | Root cause evaluation |
| Risk Matrix | Overall impact classification |
Impact Assessment
Every proposed change should undergo a comprehensive impact assessment before approval. The purpose is to identify all systems, processes, products, documents, validations, and regulatory commitments that may be affected.
Typical Areas for Impact Assessment
- Product quality
- Patient safety
- Manufacturing process
- Cleaning validation
- Process validation
- Equipment qualification
- Computer system validation
- Utilities qualification
- Analytical methods
- Specifications
- Batch records
- SOPs
- Training requirements
- Stability studies
- Regulatory submissions
- Packaging components
- Labeling
- Supplier qualification
Cross-Functional Review
Because pharmaceutical changes often affect multiple departments, cross-functional review is essential. Each function evaluates the proposed change from its area of responsibility.
| Department | Typical Responsibility |
|---|---|
| Quality Assurance | Overall compliance and approval |
| Production | Manufacturing impact assessment |
| Quality Control | Analytical impact review |
| Engineering | Equipment and utility evaluation |
| Validation | Qualification requirements |
| Regulatory Affairs | Regulatory filing assessment |
| Warehouse | Material handling implications |
| IT | Computer system assessment |
Approval Process
Only after technical evaluation and risk assessment should the proposed change be approved. Electronic approval systems are commonly used to maintain data integrity and audit trails.
Typical approvals include:
- Initiator
- Department Head
- Engineering
- Validation
- Quality Control
- Production
- Regulatory Affairs (where applicable)
- Quality Assurance (final approval)
Implementation Planning
Once approved, a detailed implementation plan is prepared to ensure the change is introduced in a controlled and traceable manner.
The implementation plan typically includes:
- Implementation schedule
- Responsible personnel
- Training plan
- Qualification activities
- Validation activities
- Documentation updates
- Material disposition plan
- Risk mitigation measures
- Contingency plan
Validation Requirements
Many pharmaceutical changes require qualification or validation activities before routine implementation.
Examples include:
- Equipment Qualification (IQ/OQ/PQ)
- Process Validation
- Cleaning Validation
- Method Validation
- Computer System Validation
- Transportation Validation
- Packaging Validation
- Utility Qualification
The extent of validation depends on the level of risk associated with the proposed change.
Documentation Updates
No change should become effective until all associated documentation has been revised, reviewed, approved, and made available to users.
Documents frequently requiring updates include:
- Standard Operating Procedures
- Batch Manufacturing Records
- Batch Packaging Records
- Specifications
- Validation Protocols
- Validation Reports
- Master Formula Records
- Equipment Logbooks
- Preventive Maintenance Procedures
- Calibration Procedures
- Training Records
Training Requirements
Employees affected by the change should be trained before implementation. Training records provide objective evidence that personnel understand the revised procedures and responsibilities.
Training may include:
- Revised SOPs
- Equipment operation
- Safety procedures
- Manufacturing instructions
- Analytical methods
- Computerized systems
- Cleaning procedures
Implementation of the Approved Change
Implementation should follow the approved action plan. Any unexpected issues encountered during execution should be documented and evaluated before continuing.
Changes should never be implemented outside the approved scope without initiating a new Change Control or formally amending the existing one.
Effectiveness Verification
Implementation alone does not complete the Change Management process. The organization should verify that the change achieved its intended objectives without introducing unintended consequences.
Typical effectiveness checks include:
- No increase in deviations
- No increase in complaints
- No adverse trend in OOS results
- Stable process capability
- Successful validation outcomes
- No regulatory compliance concerns
- Consistent product quality
Closure of Change Control
Quality Assurance reviews all supporting documentation before formally closing the Change Control. Closure should only occur after confirming that:
- All action items are completed.
- Required validations are approved.
- Training has been completed.
- Documentation has been updated.
- Effectiveness checks are satisfactory.
- Regulatory commitments have been fulfilled.
Common FDA 483 Observations Related to Change Management
Regulatory inspections frequently identify deficiencies where organizations fail to implement effective change management practices.
Examples include:
- Changes implemented without Quality Unit approval.
- Lack of documented risk assessment.
- Incomplete impact assessment.
- Failure to evaluate validation requirements.
- Implementation before SOP revision.
- Inadequate training after procedural changes.
- Failure to assess regulatory reporting requirements.
- Poor documentation supporting technical justification.
- Delayed closure of Change Controls.
- No effectiveness verification after implementation.
Common Mistakes to Avoid
- Using Change Control as a documentation exercise rather than a risk management tool.
- Classifying high-risk changes as minor to expedite approval.
- Ignoring indirect impacts on validation or regulatory commitments.
- Closing changes before completing effectiveness checks.
- Poor communication between departments.
- Implementing changes before personnel training.
- Inadequate scientific justification for proposed changes.
- Failure to update controlled documents.
Continue to Part 3: The final section covers best practices for building a robust Change Management System, practical case studies, KPIs and metrics, internal audit focus areas, frequently asked questions (FAQs), conclusion, regulatory references, and a regulatory disclaimer suitable for publication on PharmaShare.
Regulatory Disclaimer
Disclaimer: This article is intended solely for educational and informational purposes. While every effort has been made to ensure accuracy based on current pharmaceutical regulations and industry best practices, readers should always refer to the latest official guidance issued by regulatory authorities such as the US FDA, EMA, MHRA, WHO, PIC/S, and ICH before making compliance or business decisions. Company-specific Standard Operating Procedures (SOPs), internal quality systems, and applicable national regulations should always take precedence where relevant.
Author: Mahummed Asif
Founder, PharmaShare | Pharmaceutical Quality & Regulatory Professional