Types of Variation Filing in USA for Pharma: PAS, CBE-30, CBE-0 & Annual Report Explained
Last Updated: April 2026 | For: Regulatory Affairs, CMC, QA Professionals
If you work in Regulatory Affairs for US FDA submissions, you face this question in every CMC meeting: “Is this change a PAS or CBE?”
Choose wrong = FDA delay + 483 observation + product shortage. US FDA doesn’t use EMA’s “Type IA/IB/II” system. For NDA, ANDA, BLA holders, post-approval changes are governed by 21 CFR 314.70 + 21 CFR 601.12.
This guide breaks down all 4 FDA variation filing types with real examples so your RA team files right the first time.
1. Why US FDA Has 4 Variation Categories?
FDA’s logic is simple: Risk = Review Time.
- High risk to safety/efficacy → FDA must review before implementation
- Low risk → Company can implement + inform FDA later
Goal: Protect patient safety while giving manufacturers flexibility for continuous improvement.
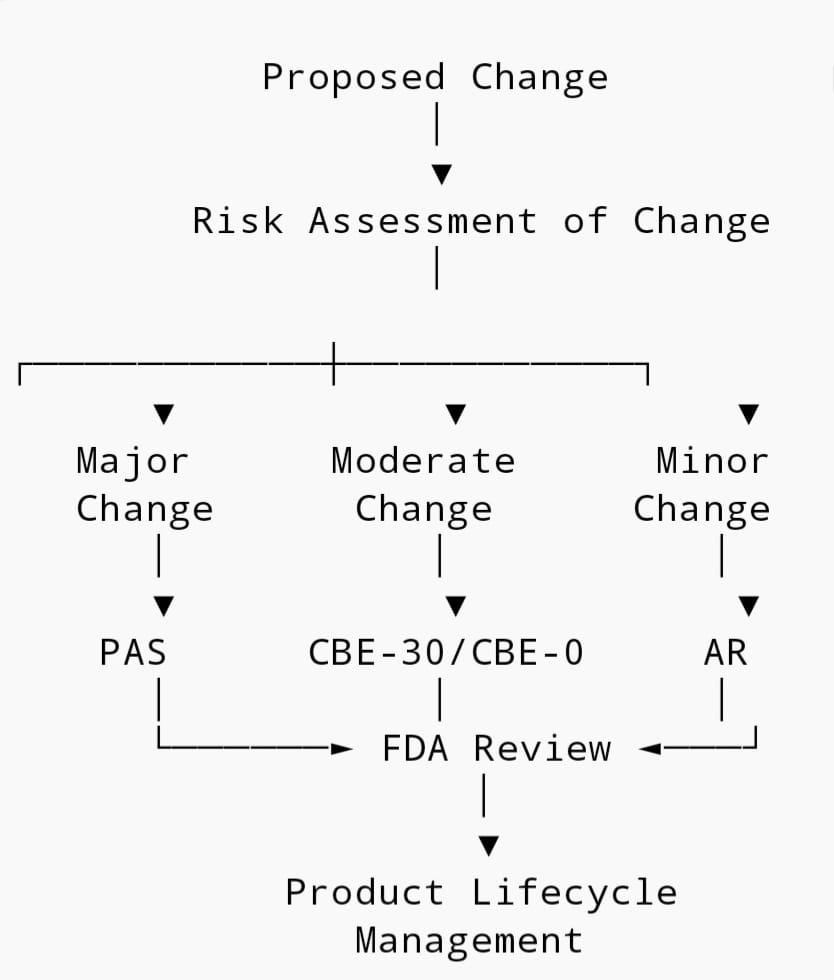
2. The 4 Types of US FDA Post-Approval Changes
Type 1: Prior Approval Supplement – PAS
Regulation: 21 CFR 314.70(b) for drugs, 21 CFR 601.12(b) for biologics
Risk Level: Highest – Substantial potential to affect safety, identity, strength, quality, purity, or potency
When to file PAS:
- Change in API manufacturing site with different synthesis route
- New container closure system for sterile injectables
- Change in sterilization method from steam to gamma
- Tightening or widening of specification limits
- Addition of new strength/dosage form
Critical Rule: Cannot implement until FDA approval letter is received. Early implementation = “Distribution of unapproved product” = Warning Letter.
Real Example: Moving API manufacturing from Hyderabad Site A to new Visakhapatnam Site B = PAS
Type 2: Changes Being Effected in 30 Days – CBE-30
Regulation: 21 CFR 314.70(c), 21 CFR 601.12(c)
Risk Level: Moderate – Moderate potential to affect product quality
When to file CBE-30:
- Change in-process controls within approved ranges
- New analytical method with full validation data
- Addition of alternate API supplier already in approved DMF
- Minor batch formula change within approved range
- Equipment change of same design/operating principle
Real Example: Changing HPLC column from Waters XBridge to Agilent Zorbax with same C18 chemistry = CBE-30
Type 3: Changes Being Effected – CBE-0
Regulation: 21 CFR 314.70(d), 21 CFR 601.12(d)
Risk Level: Low – Minimal potential to affect product quality
When to file CBE-0:
- Editorial labeling changes per FDA guidance
- Change in pack count within approved range, e.g. 10s to 30s
- Minor equipment change with documented no-impact
- Addition of alternate excipient supplier
Real Example: Changing carton artwork color from Pantone 185C to 186C with no text change = CBE-0
Type 4: Annual Report – AR
Regulation: 21 CFR 314.70(e), 21 CFR 601.12(e)
Risk Level: Lowest – Changes with minimal risk listed in guidance
When to file AR:
- Minor labeling change like adding distributor address
- Change in batch number format
- Annual stability data update
- Change in non-contact packaging component supplier
Risk: Forgetting to include change = FDA 483 “Unreported post-approval change”.
Real Example: Adding “Manufactured for ABC Pharma, USA” line below manufacturer address = Annual Report
3. PAS vs CBE-30 vs CBE-0 vs AR: Decision Matrix
| Question to Ask | If YES → File This | FDA Approval Needed? | Implementation Timeline |
|---|---|---|---|
| Does change affect sterilization, impurity profile, or route of synthesis? | Prior Approval Supplement | Yes | After FDA approval letter |
| Does change need new validation but no new safety data? | CBE-30 | No | After 30 days, if no FDA hold |
| Is it editorial/labeling change with zero quality impact? | CBE-0 | No | Immediately upon filing |
| Is it listed as “Annual Reportable” in FDA guidance? | Annual Report | No | Report in yearly AR |
Golden Rule: When in doubt, file higher category. FDA prefers PAS over a 483 for under-reporting.
4. 3 Costly Mistakes RA Teams Make in USA
- Treating CBE-30 as CBE-0: “We validated new HPLC method internally, so low risk”. FDA view: Any new analytical method = CBE-30 minimum because method impacts release data quality.
- Implementing PAS before approval: Production pressure to avoid stockout. FDA calls this adulterated product under 501(a)(5) = Import Alert + Warning Letter.
- Forgetting AR: Small changes like primary packaging paper GSM change get missed. FDA sees during next PAI → “Failure to report changes” observation.
5. FDA Expectations in 2026
- PACMP – Post-Approval Change Management Protocol: If approved in original NDA, you can downgrade PAS to CBE-30. Submit protocol upfront to save time later.
- Comparability Protocol: For biotech BLAs, pre-approved protocol allows faster CBE instead of PAS for process changes.
- eCTD Mandatory: All supplements must be in eCTD format 3.2.2. Paper submissions rejected since May 2018.
- QbD Linkage: Changes within approved design space may qualify as CBE-30 or AR instead of PAS.
Key Takeaway for RA Professionals
US FDA variation system has only 4 buckets vs EMA’s 20+ types. But “judgment” decides the bucket. Wrong categorization = 483 + recall risk + loss of FDA trust.
Before any CMC change, your RA + QA + Technical team should answer 3 questions together:
- Does it impact patient safety or efficacy? → PAS
- Does it impact quality data/method? → CBE-30
- Does it impact nothing? → CBE-0 or AR
Maintain a “Change Categorization SOP” with 20+ real examples from your own portfolio. It saves 100+ hours of FDA information requests per year.
Disclaimer: This article is for educational purpose only. Final filing category must be decided by your company’s RA head based on product-specific data and FDA guidance.
Read Next: FDA 483 Observations on CAPA and CAPA Effectiveness

About the Author
Mahummed Asif is a pharmaceutical QA professional with 16 years experience in GMP, QMS, Product Complaint Management, Change control, risk management, and USFDA audit preparation.